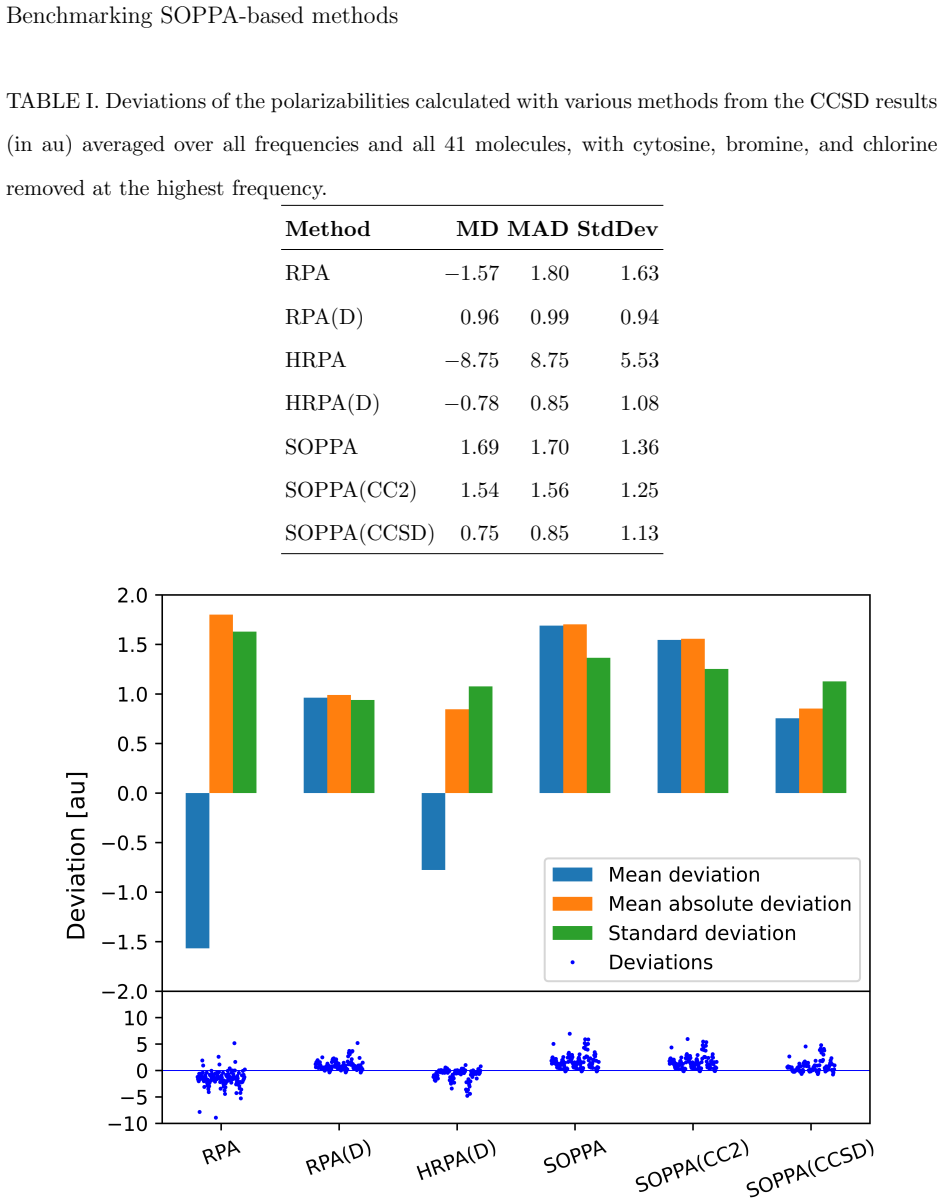
7
Ozone double ionization yields excited oxygen ions
Single-Photon Double Ionization of Ozone
First valence double-ionization spectrum and calculations show both ground and excited O+ fragments, revealing more breakup pathways than此前已
 full image
full image
abstract click to expand
Ozone (O3) is a triatomic molecule of central importance in the chemistry and physics of the Earth's and other planetary atmospheres. Beyond its environmental significance, a detailed understanding of the electronic structure and ionization dynamics of ozone is essential for modeling atmospheric, ionospheric, and astrochemical processes. In the present work, we substantially extend the experimental and theoretical characterization of ozone into the regime of valence double photoionization. Using HeII-alpha, HeII-beta, and higher-energy vacuum ultraviolet radiation in combination with a versatile multiple charged-particle correlation detection technique, we report the first single-photon valence double ionization electron spectrum of O3. To interpret the experimental observations, we mapped the lowest potential energy surfaces of dicationic ozone employing post-Hartree-Fock multi-configurational-interaction methods, and computed with high accuracy the energetics of the relevant dissociation channels. Our results demonstrate that dissociative double ionization of ozone produces electronically excited cationic atomic oxygen fragments in addition to the ground-state dissociation pathway, revealing a richer fragmentation dynamics than hitherto recognized.