1
Degree and distance contact rules slow epidemic growth
Even mild dependencies shift spread from explosive to polynomial rates on networks with geometry.
 full image
full image
abstract click to expand
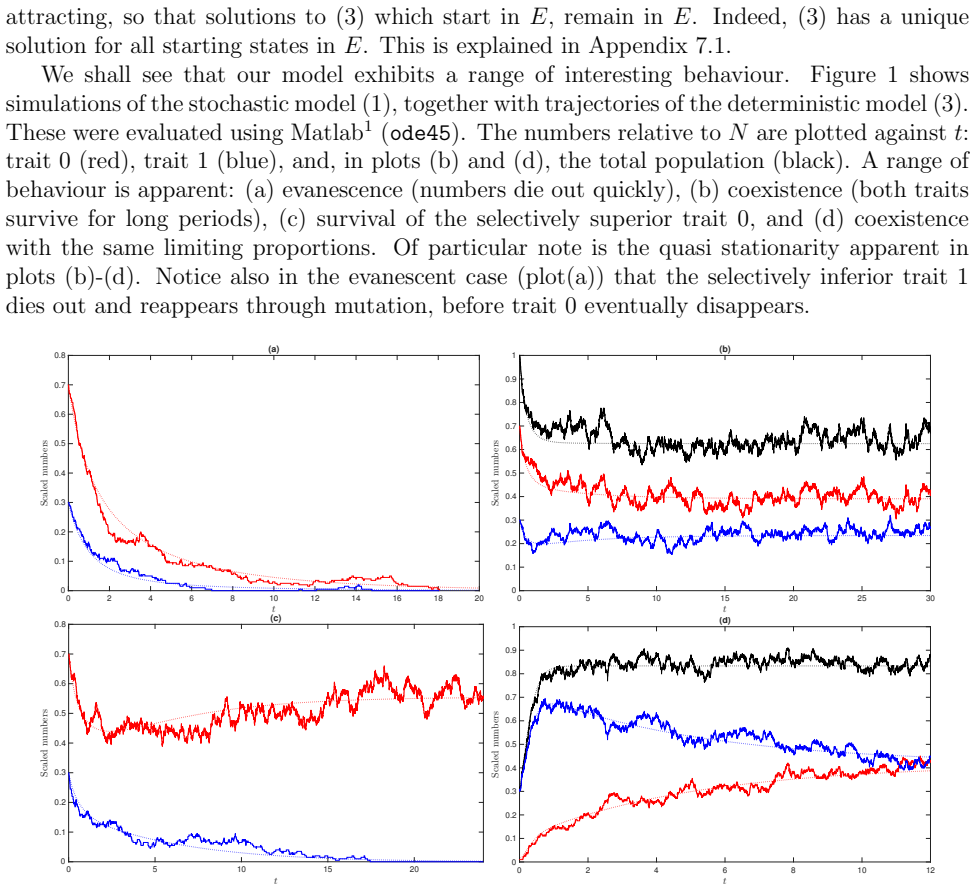
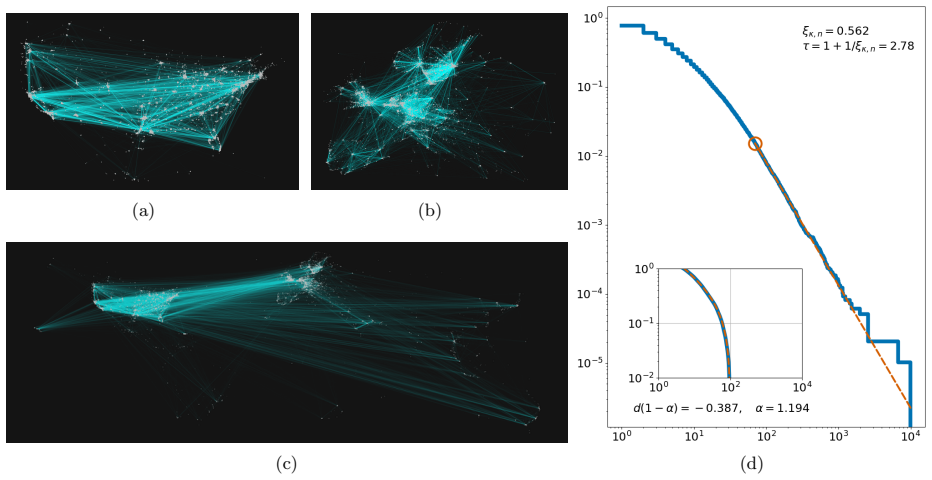
It is a fundamental question in epidemiology to estimate, model and predict the growth rate of a pandemic. Analogously, analysing the diffusion of innovation, (fake) news, memes, and rumours is of key importance in the social sciences. The resulting epidemic growth curves can be classified according to their growth rates. These have been found to range from exponential to both faster super-exponential curves and slower subexponential or polynomial curves. Previous research has lacked a unified explanatory framework capable of accommodating super-exponential, (stretched) exponential, and polynomial growth patterns within the same contact network. In this paper we propose a simple agent-based network model that can capture all these phases. We provide such a framework by modelling how transmission rates depend on spatial distance and on individuals' numbers of contacts. By comparing the growth rate of spreading processes with or without degree-dependent and/or distance-dependent contact rates through data-driven and synthetic simulations on real and modelled networks with underlying geometry, we find evidence that even a 'sublinear presence' of these causes may cause a significant slow down of the growth rate on the same underlying network. We find that the growth rate is governed by a combination of three factors: geometry, the prevalence of weak ties, and superspreaders. We confirm our results with rigorous proofs in a theoretical model, using a spatial multiscale-argument in long-range heterogeneous first passage percolation. Our results give a plausible explanation of why the consecutive waves of a single pandemic can differ in their growth even if their spreading mechanisms are similar.