0
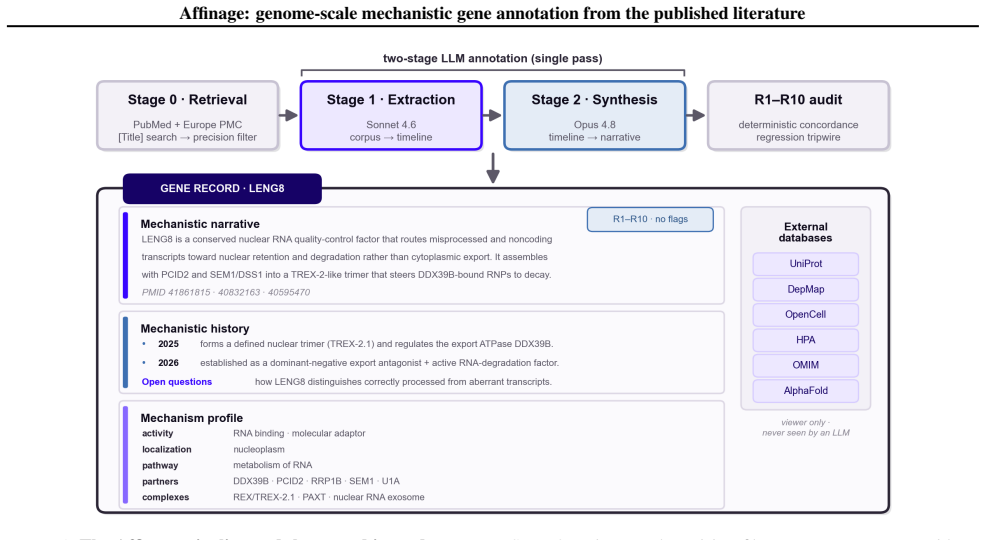
Pipeline annotates mechanisms for 19,293 human genes from papers
Affinage: genome-scale mechanistic gene annotation from the published literature
Affinage pulls direct experimental evidence to fill gaps where UniProt entries are empty or minimal.
 full image
full image
abstract click to expand
Understanding the mechanistic function of a gene is a critical starting point for biology. However, for much of the human proteome that knowledge is scattered across thousands of primary papers or remains poorly established, while the curated databases biologists rely on can lag years behind recent literature. Large language models can now read and synthesize that literature on demand, but doing so faithfully for many genes is an expensive, non-reproducible retrieval session that does not scale across users. Here, we present Affinage, an LLM pipeline that performs this retrieval and mechanistic reasoning once per gene--from the primary literature alone--and stores the result as a reusable, structured annotation. A biologist-designed reading pass extracts only direct experimental evidence, and a synthesis pass reasons over those findings alone. Applied across the genome, Affinage annotates 19,293 human protein-coding genes. This analysis provides mechanism for thousands of genes whose UniProt function is empty or a stub, beating the curated reference on 99.1% of head-to-head genes as scored by a cross-family LLM judge. Affinage also delineates the 10% of the proteome that remains mechanistically uncharacterized and will serve as a continuously-updated, literature-grounded census of gene function. All records are released openly at https://affinage.wi.mit.edu . More broadly, Affinage serves as an example of how domain experts can encode their expertise into scalable LLM pipelines to improve the publicly available data that guides biological hypotheses and experimentation.