0
Phosphorylation model admits regime with two stable equilibria
Thermodynamic Limits of Stochastic Chemical Reaction Networks with Phosphorylation
With substrate fixed at N and enzymes scaling with N, specific catalytic constants yield three equilibria of which two are stable.
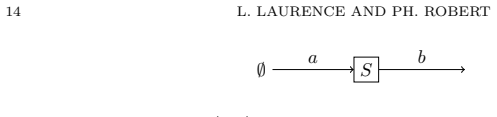 full image
full image
abstract click to expand
In this paper we investigate the stability properties of a fundamental mechanism of biological cells called phosphorylation. The system is a chemical reaction network (CRN) for which a chemical species, {\em the substrate}, can be sequentially transformed into two phosphorylated forms, by the activity of two types of enzymes, one type for phosphorylation, the other for dephosphorylation. We investigate a stochastic representation of this model, under the mass action kinetics. The total mass of the substrate is fixed at $N$, while the total mass of enzymes scales proportionally to $N$. The asymptotic behavior, when $N$ is large, of the concentrations of all chemical species is studied.
We investigate the possible {\em stable} subsets of chemical species for the kinetics of the law of mass action. A stable subset is such that, with a convenient initial state, the number of copies of the species of this subset remains $O(1)$ on any finite time interval as $N$ gets large. The role of the twelve reaction rate constants, {\em the catalytic constants} of the CRN, is investigated from this point of view. An averaging principle of the corresponding Markov process is established for several regimes of the CRN. It is shown in particular that there exists a regime with three equilibrium points, with two of them stable. The proofs of the results rely on stochastic calculus with Poisson processes, convenient couplings of subsets of coordinates of the Markov process, technical results on $M/M/\infty$ queues, and a stability analysis of a dynamical system in $\mathbb{R}_+^4$.

































































